The main interests of the lab can be divided into 4 main areas:
RNP dynamics along the mRNA during normal and misregulated translation
Tight control of cytoplasmic gene expression is orchestrated by the action of multiple ribonucleoprotein (RNP) complexes. One prominent example is the human ribosome, which undergoes drastic changes as it scans through the mRNA and engages in protein synthesis. In recent years, translation profiling (with techniques such as ribosome profiling, or Ribo-seq) has exposed a previously under-appreciated complexity in translation with incredible resolution, revealing active translation on the non-coding transcriptome, and allowing for signal processing investigation of ribosome dynamics.
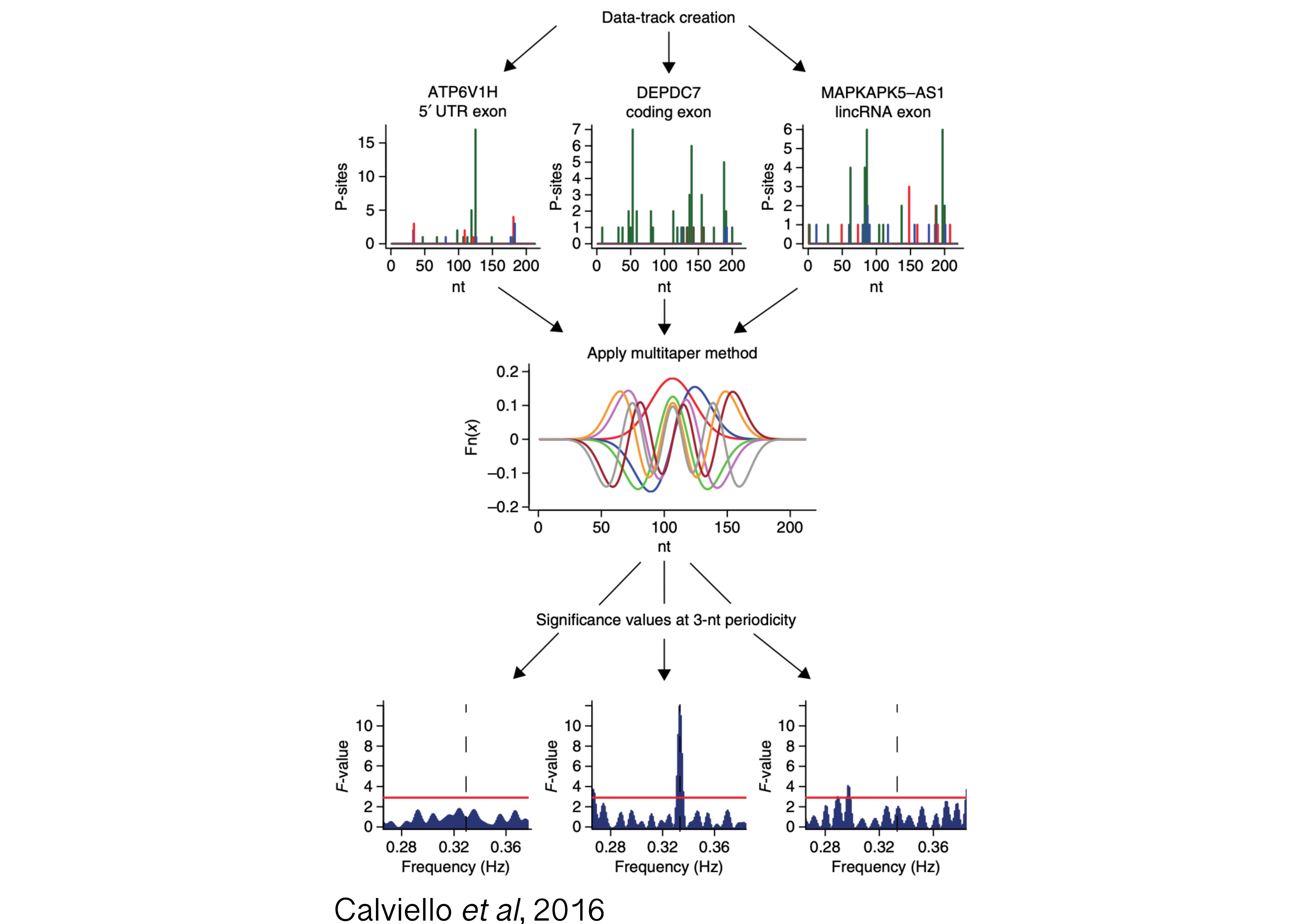
Moreover, analysis of protein-RNA complexes revealed an unexpected diversity to the ribosomal RNP itself, with hundreds of factors interacting with the ribosome throughout the translation cycle. However, our ability to characterize binding and functions of heterogeneous ribosomal complexes along the transcriptome remains limited.
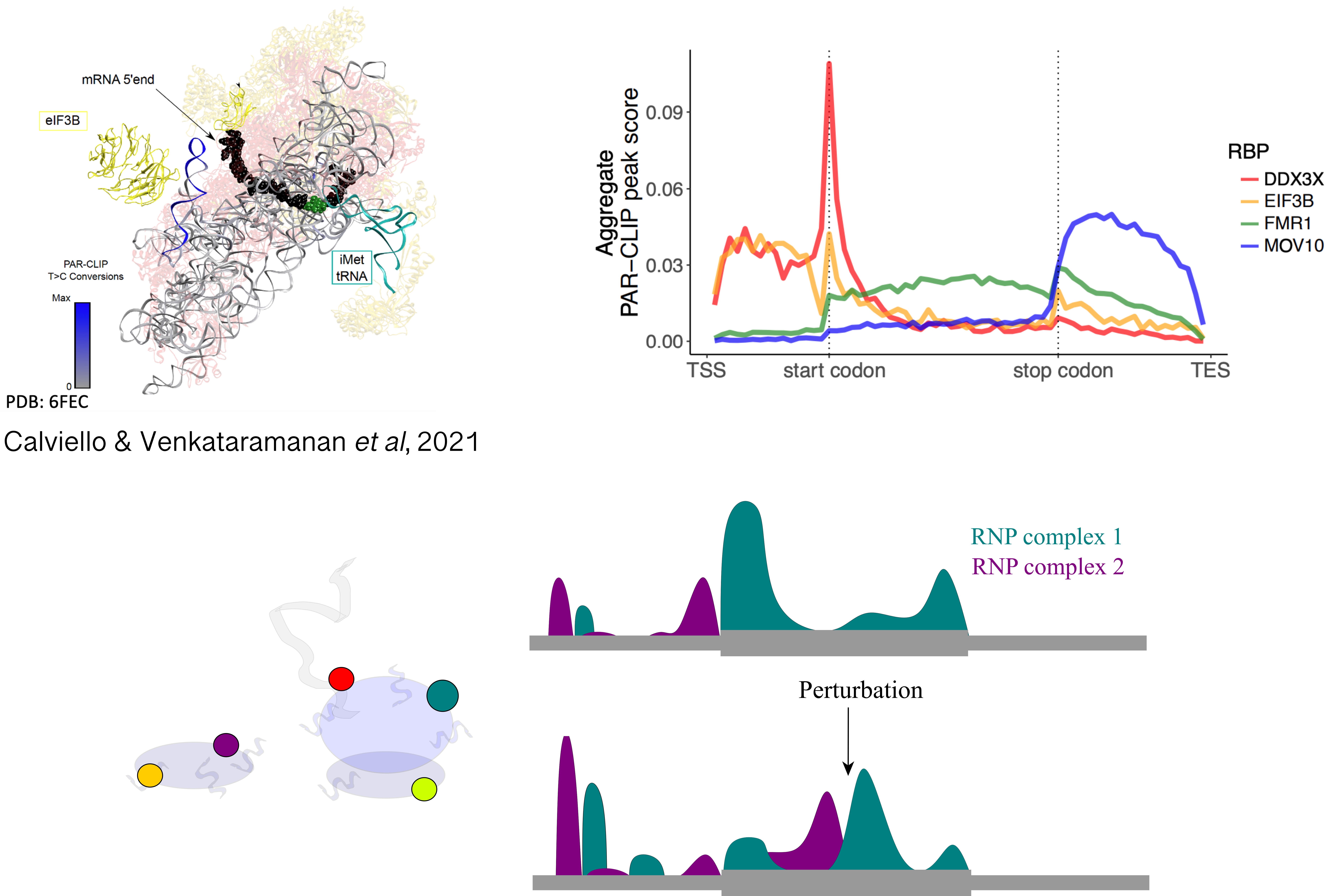
Projects in this area aim at quantifying the interplay between functions of heterogeneous RNPs and RNA sequence-structure features, using experimental methods aimed at selective profiling of different RNP complexes, and computational analysis of transcriptome-wide tracks aided by signal processing and deep learning techniques. In addition, we aim at robustly detecting alternative modes of translation initiation and elongation, often ignored by standard analysis methods.
Translation in gene expression control across cell types and states
RNA cis-regulatory elements and RBP binding can modulate the rate of protein synthesis on the mRNA, providing an additional mode of gene expression control. Moreover, decades of detailed molecular investigations in protein synthesis have uncovered multiple connections between nascent RNA processing, translation, and mRNA decay. Despite the vast amounts of data depicting these phenomena transcriptome-wide and with great accuracy, an analytical framework able to quantify and predict regulation at different steps of the gene expression cascade is missing.
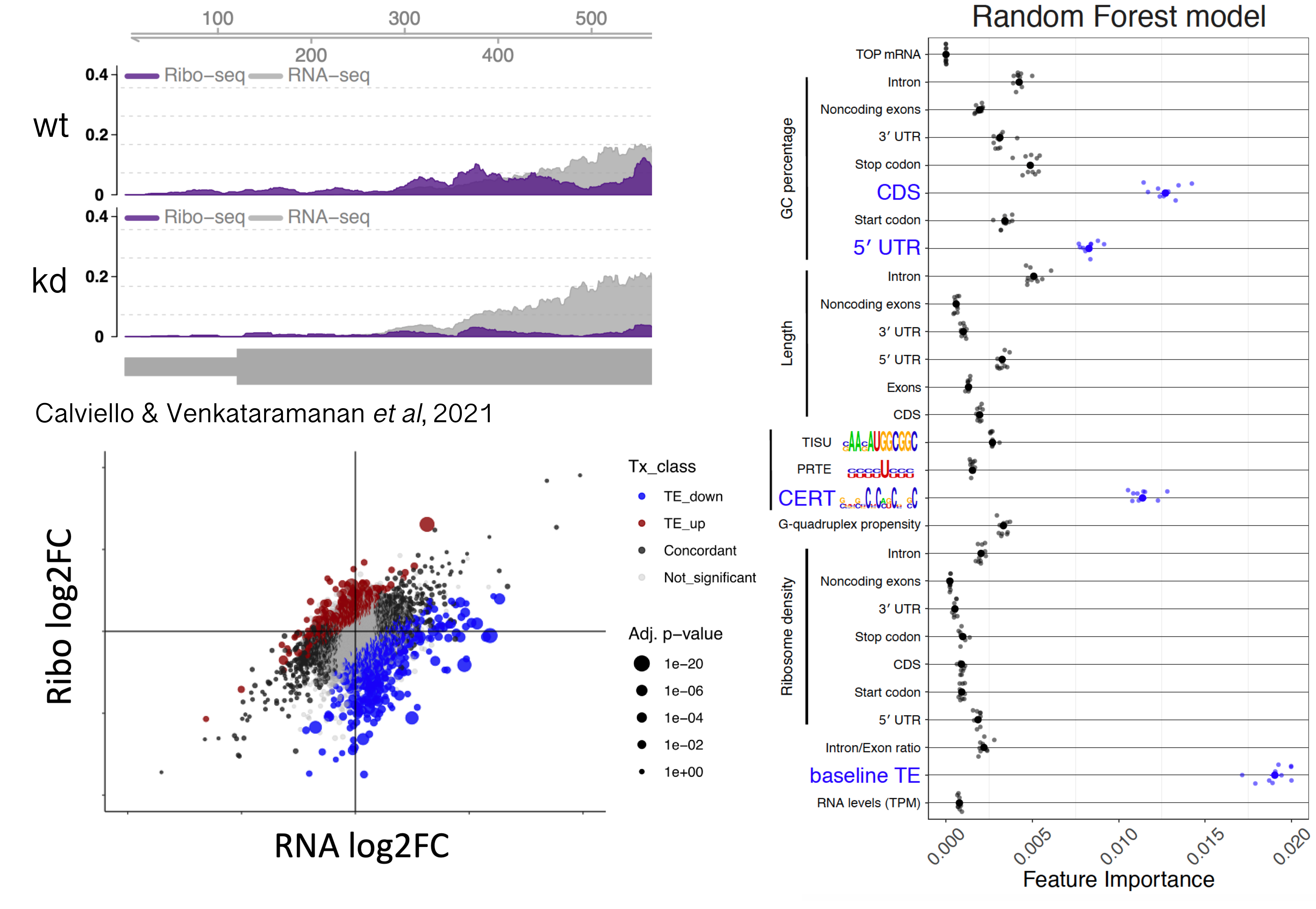
Projects in this area will make extensive use of thousands of available Ribo-seq and RNA-seq datasets, controlled in silico experiments and machine learning to quantify different modes of gene expression regulation in multiple cell types and under different perturbations. In addition, more targeted studies in this area will focus on elucidating the molecular functions of RNA binding proteins, subcellular regulatory processes, and regulation by signaling pathways like mTOR and the Integrated Stress Response.
Alternative RNA processing and its contribution to protein synthesis and cell physiology
RNA-sequencing (RNA-seq) data has enabled researchers to quantify expression of thousands of genes, from more canonical example of protein coding genes to cryptic transcripts of unknown function; moreover, different transcript isoforms from the same genes have often been shown to exhibit tissue-specific functions, highlighting the need for transcript-level investigation into gene regulation. For hundreds of genes, computational analysis of Ribo-seq data has provided evidence for translation and also co-translational mRNA decay in a transcript-specific fashion, providing a powerful window into the cytoplasmic fates of thousands of transcripts. However, the extent of transcript-specific translational control, and the physiological relevance for the presence of multiple translated isoforms remains elusive.
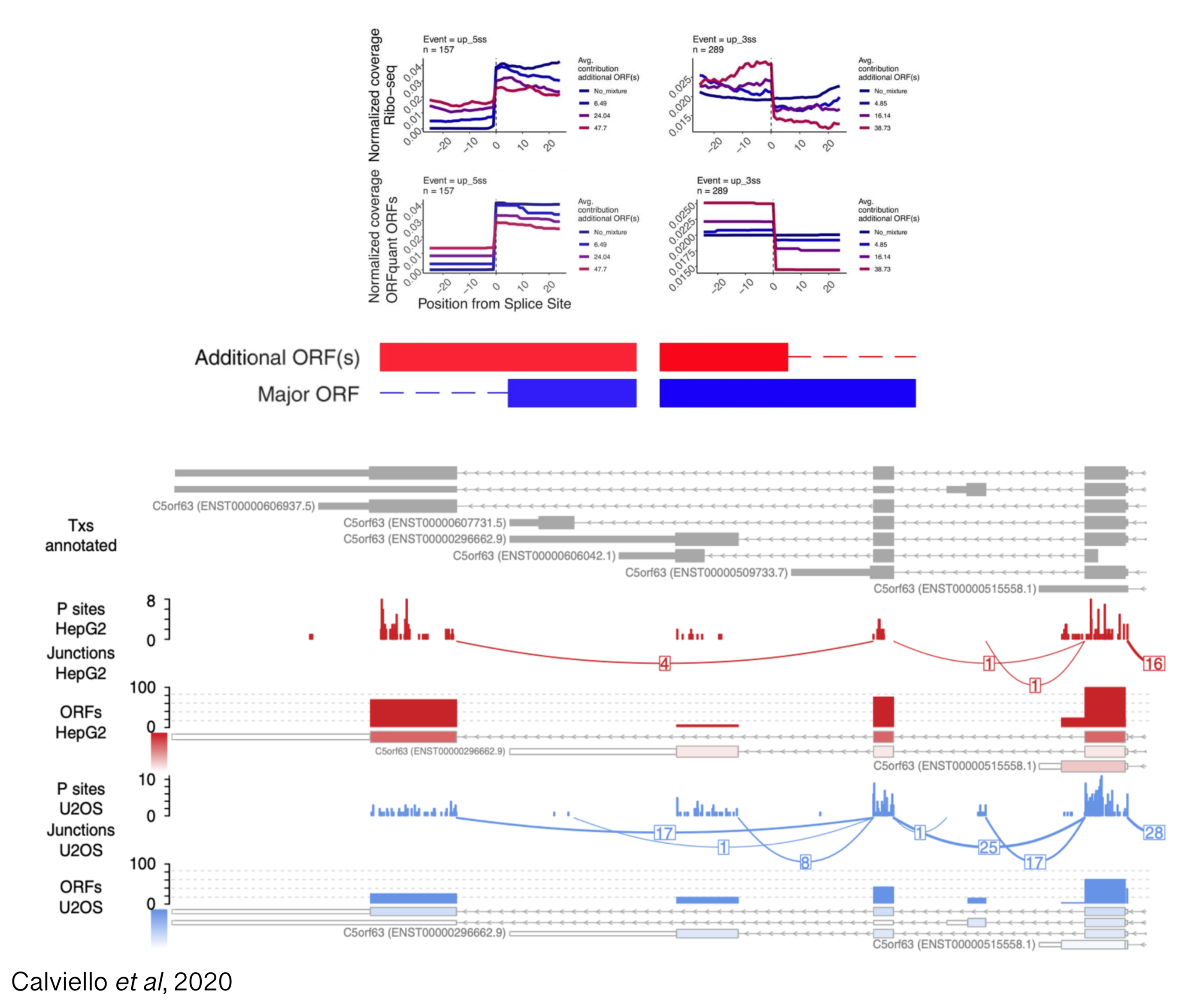
Projects in this area will aim at quantifying the contribution of annotated and novel transcripts to the ensemble of translated sequences, using experimental methods measuring translation and RNA decay (with a focus on long-read technologies), and computational strategies to identify novel transcripts and deconvolve signals coming from multiple isoforms.
In addition, in collaboration with the Neurogenomics department, we will investigate how the interplay of alternative splicing, co-translational RNA surveillance and localized translation regulation shapes neuronal functions and underlies several neuronal pathologies.
Proteogenomics applications between RNA and protein
RNA sequencing technologies are essential to detect expressed transcripts, thus providing useful priors to identify synthesized proteins. As shown by different studies, careful analysis of Ribo-seq profiles can reveal protein synthesis events with high confidence and aid the discovery of new proteins being translated from uORFs, lncRNAs and alternative transcript isoforms. While earlier studies took advantage of proteomics data to qualitatively validate protein synthesis at uncharacterized loci, re-analysis of mass-spectrometry datasets revealed strong quantitative agreements between translation output and steady-state protein abundance. Despite these recent promising results, integrating transcriptomics with the complex world of proteome dynamics, while considering regulation via post-translational modifications, presence of protein isoforms, and protein subcellular localization and function, remains an unmet challenge in the post-genomic era. In addition, a complex interplay between translation elongation dynamics and co-translational protein folding ensure correct protein stability and function, confirming the importance of jointly investigating RNP dynamics and protein activity.
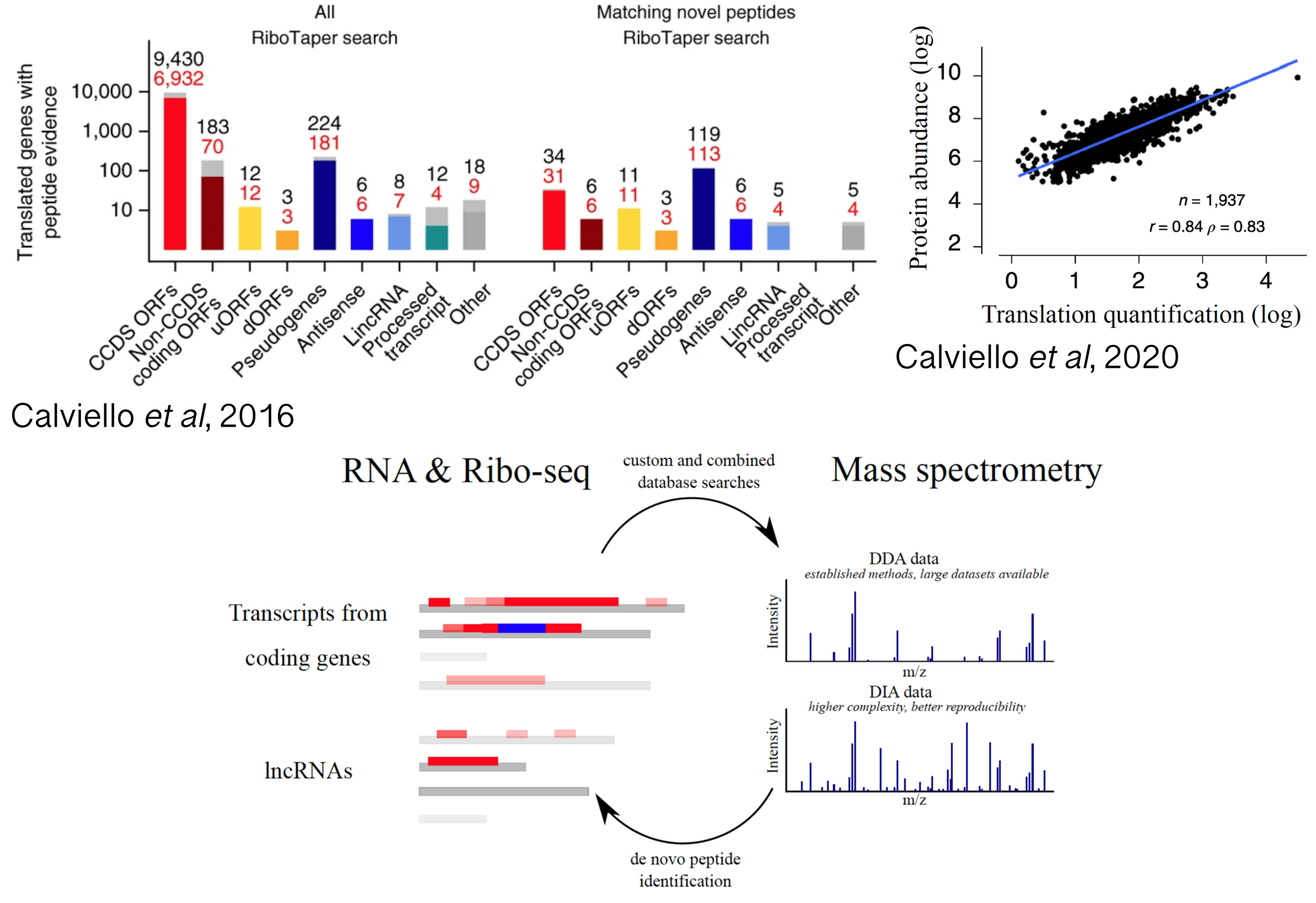
Projects in this area will analyze mass spectrometry data aided by RNA-seq and Ribo-seq analysis, with a focus on detecting and quantifying alternative proteins from translation of multiple transcripts isoforms, novel ORFs with unclear coding potential, and variations of “canonical” ORFs. In addition, we would like to initiate collaborative proteogenomics initiatives with the larger scientific community, to generate joint projects aimed at bridging next-generation sequencing and shotgun proteomics technologies.